
 Skip to navigation
Skip to navigation
Site Primary Navigation:
- About SDSC
- Services
- Support
- Research & Development
- Education & Training
- News & Events
Search The Site:
Published 11/01/2006
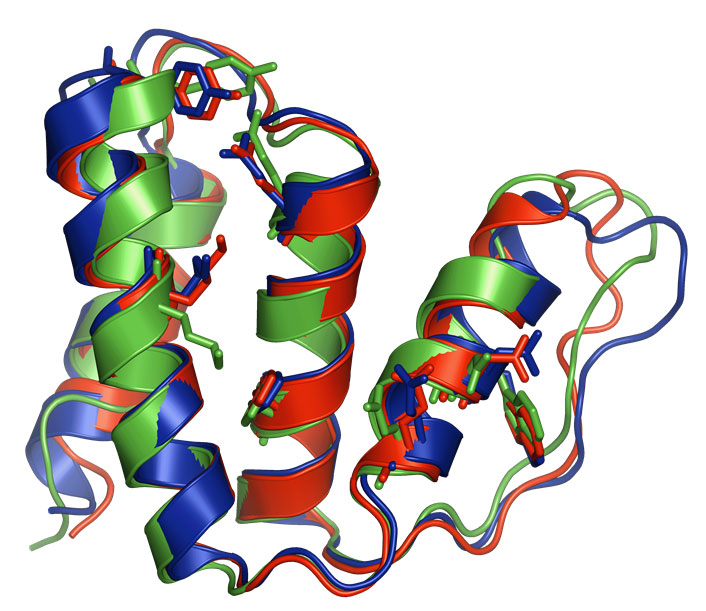
Results from a 40,000 processor run on BG/L to predict the structure of CASP target in the 7th competition. Red is the native X-ray structure, blue is the Rosetta prediction and green a low resolution NMR model. The entire prediction was completed in under six hours with the computational component taking three hours.
by Paul Mueller, UCSD
The high-stakes task of designing new drugs relies on high-tech tools, especially computerized predictions of the 3-D structure of proteins. Researchers at the San Diego Supercomputer Center (SDSC) at UC San Diego, contributing their massive computational capabilities to a collaboration with colleagues at the University of Washington (UW) and IBM, have helped to achieve the largest-ever protein structure prediction - and completed the complex simulation in less than three hours, a task that previously took weeks.
The groundbreaking demonstration, part of the biennial Critical Assessment of Structure Prediction (CASP) competition, used UW professor David Baker's Rosetta Code and ran on more than 40,000 central processing units (CPUs) of IBM's Blue Gene Watson Supercomputer, using the experience gained on the Blue Gene Data system installed at SDSC.
When Baker, who also serves as a principal investigator for Howard Hughes Medical Institute, originally developed the code, it had to be run in serial - broken into manageable amounts of data, with each portion calculated in series, one after another.
Through a research collaboration, SDSC's expertise and supercomputing resources helped modify the Rosetta code to run in parallel on SDSC's massive supercomputers, dramatically speeding processing, and providing a testing ground for running the code on the world's fastest non-classified computer.
During the CASP competition, the results of trillions of calculations a second were fed across the TeraGrid network - part of a national collaborative grid computing facility - from SDSC to Baker and his team, giving them far more computing power than had ever been available in previous CASP simulations.
The Strategic Applications Collaboration (SAC) staff at SDSC initiated the work with Baker and the codes, introducing them to NSF supercomputing resources, and helped his team scale their codes to run on supercomputers.
Ross Walker, a SAC computational scientist, managed the Baker group's access to SDSC machines during the competition, helping them optimize the code for the SDSC computers. He also provided customized resource allocation at SDSC - key to running the time-sensitive calculations as quickly as possible.
"The Blue Gene system is ideal for running protein structure prediction," said Walker, "as the process involves predicting many, many structures, optimizing them, and then scoring them, with the ultimate aim that the best scoring structure - the lowest energy - is the native structure. The more predictions we can do in a given time, the better."
CASP calculations are essentially competitions among scientists to predict protein structures and are, therefore, "very time-sensitive," said Walker. "Previously, with only limited computer power, the Baker team had to skimp both on the amount of computing they did, and on the amount of analysis they did."
Access to SDSC's supercomputing facilities was vital to the Baker group, as it allowed them both to increase the complexity of their calculations and to gain time for analysis before submitting their prediction.
"The post-production data analysis is as important as generating the models themselves," said Vatsan Raman of Baker's team. "Similarly, the experience gained on the SDSC machines was invaluable when it came to running the code on over 40,000 processors. Being able to run on the full machine at IBM was a real breakthrough for the Rosetta team, allowing us to run more complex calculations than we had attempted before, in a fraction of the time."
The team was able to generate about 120,000 structure predictions, greatly enhancing the probability of locating the lowest energy conformation, and immediately analyze those predictions, submitting the best structure as their CASP prediction.
Most noteworthy, Walker says, is that "all this was done within a day, with the calculation taking less than three hours. Run in serial, or on small clusters, as has been done heretofore, the process took many, many weeks."